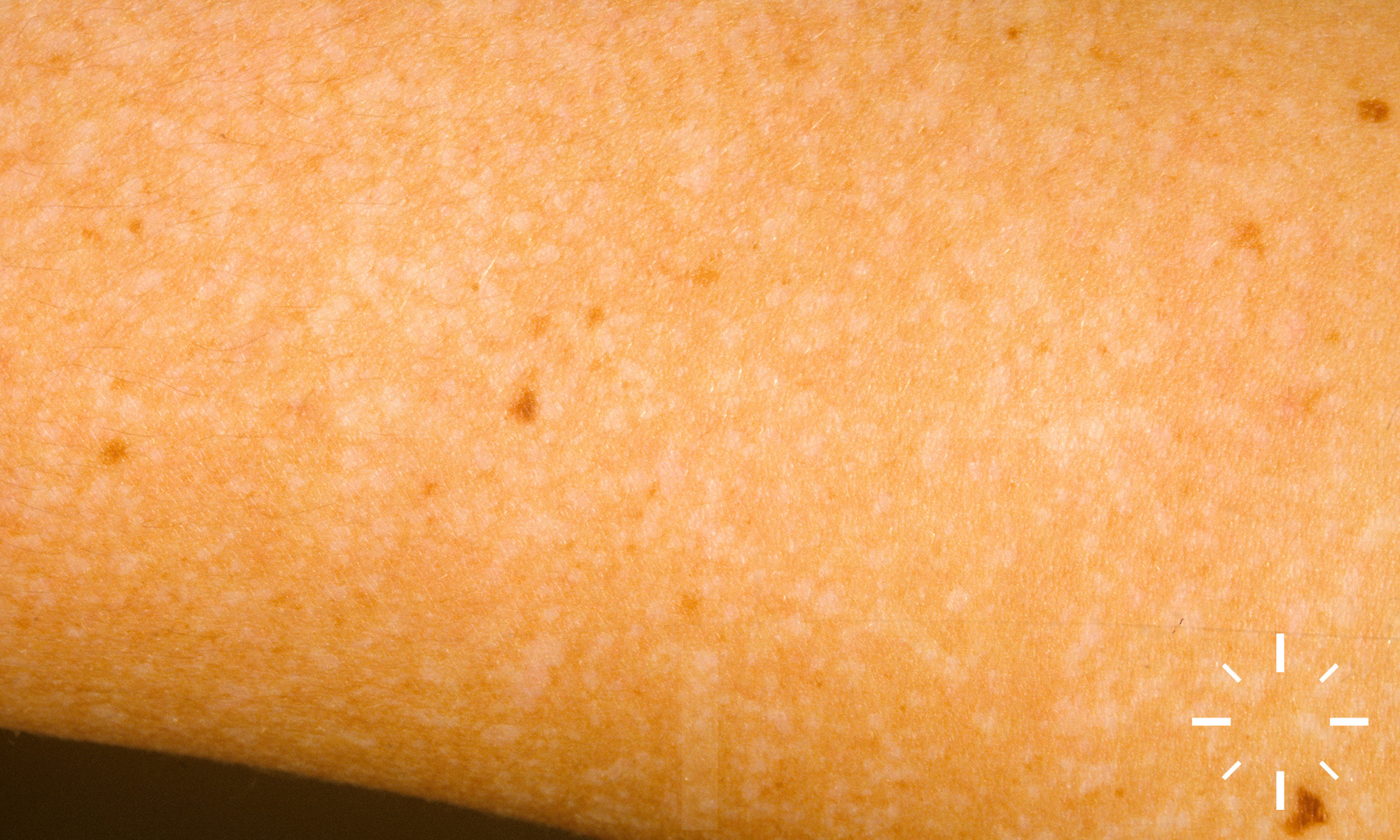
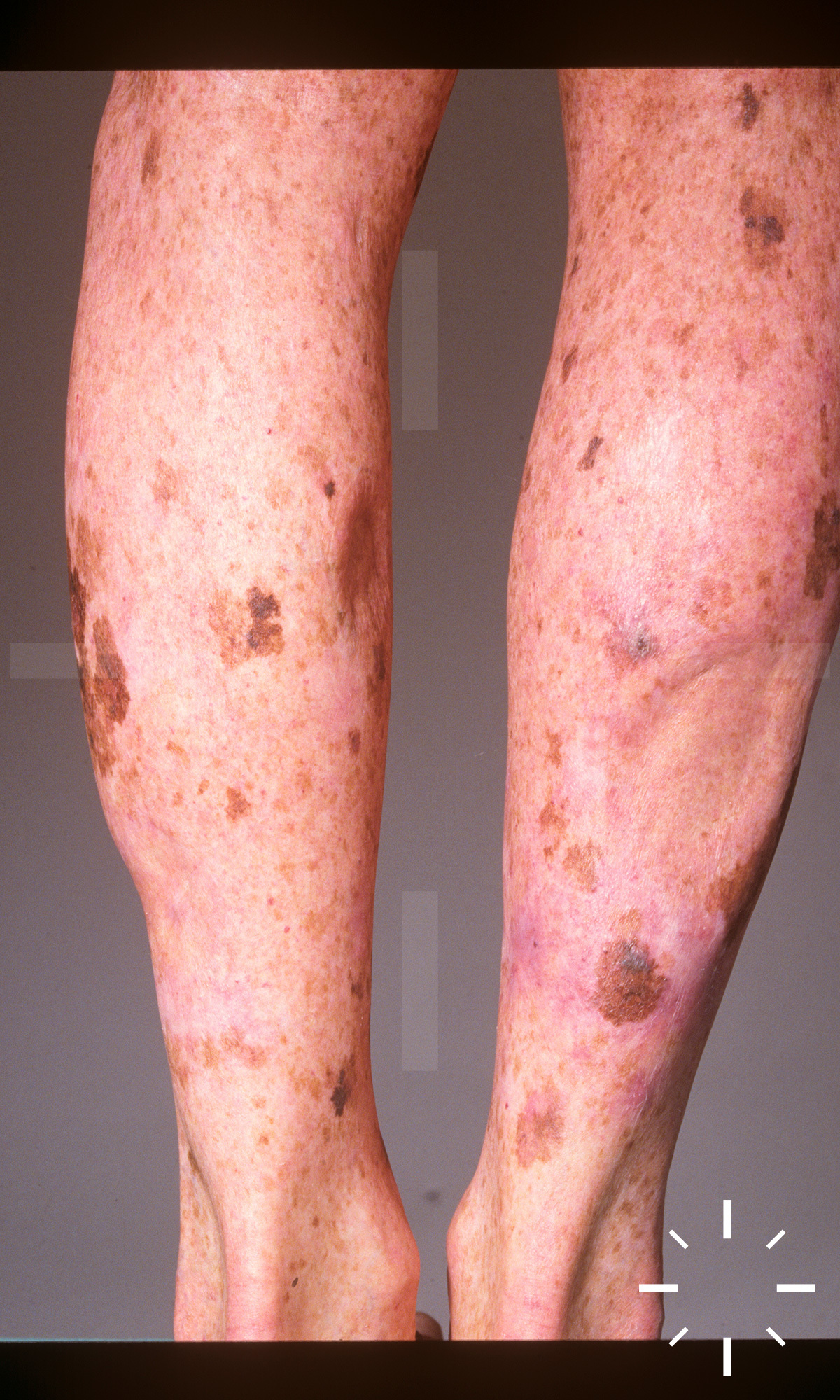
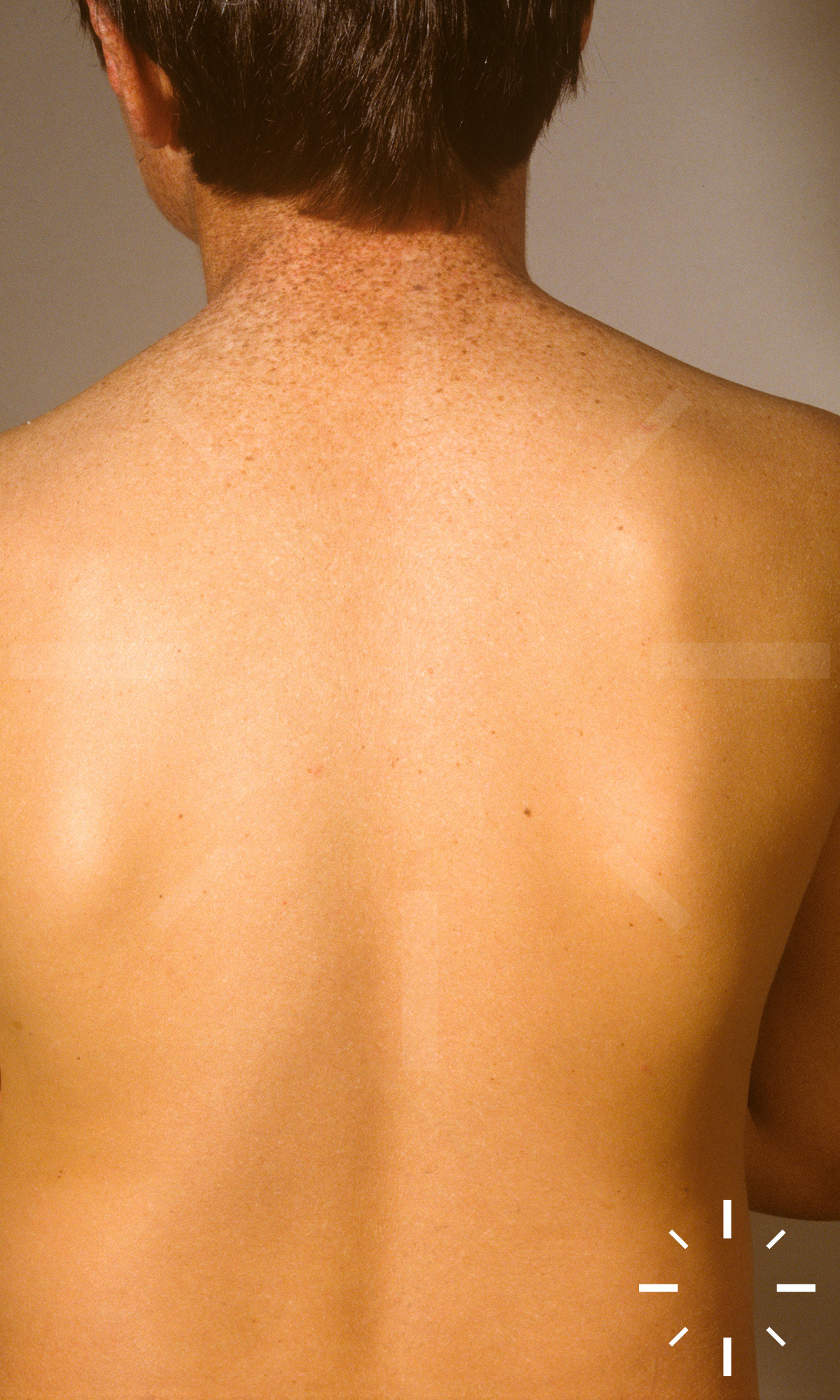
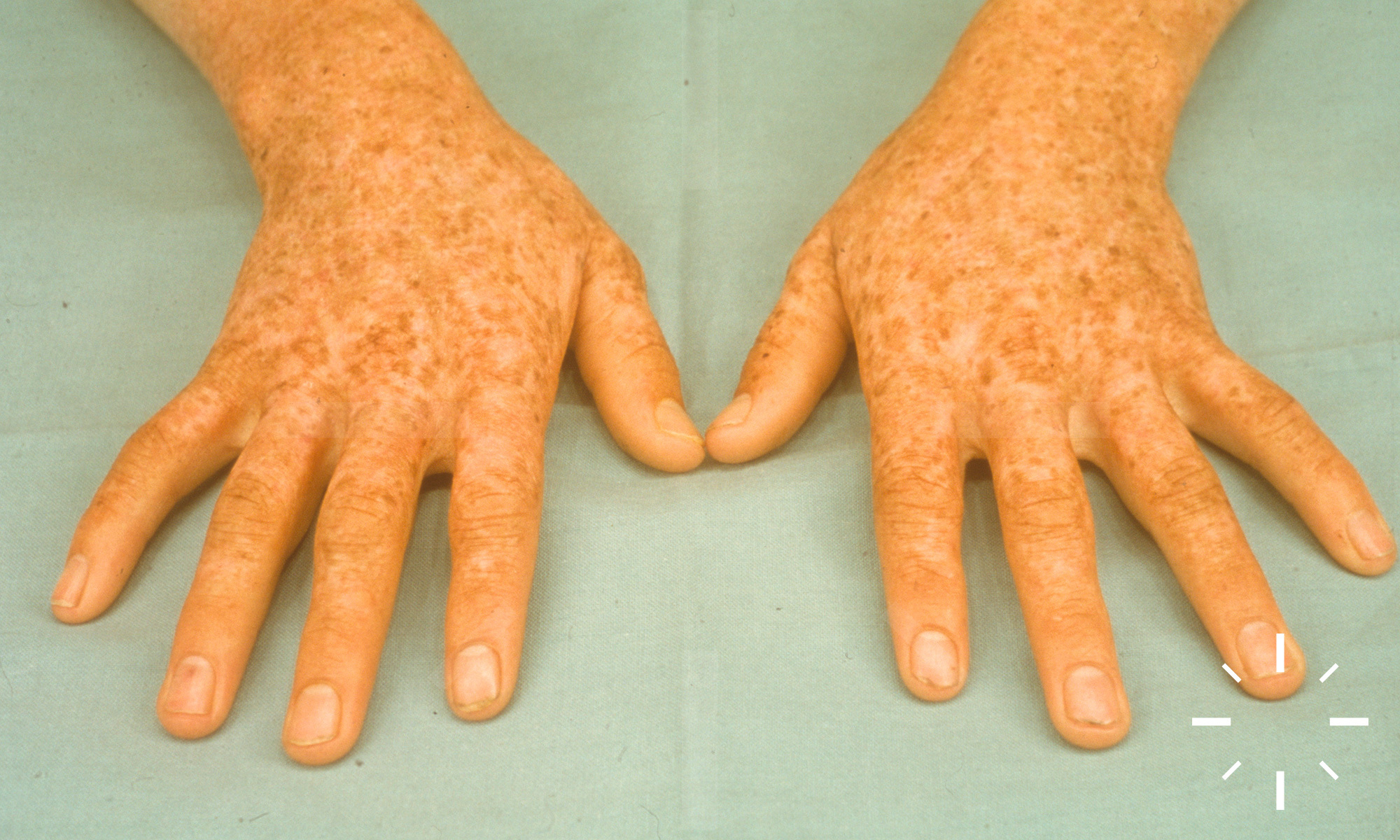
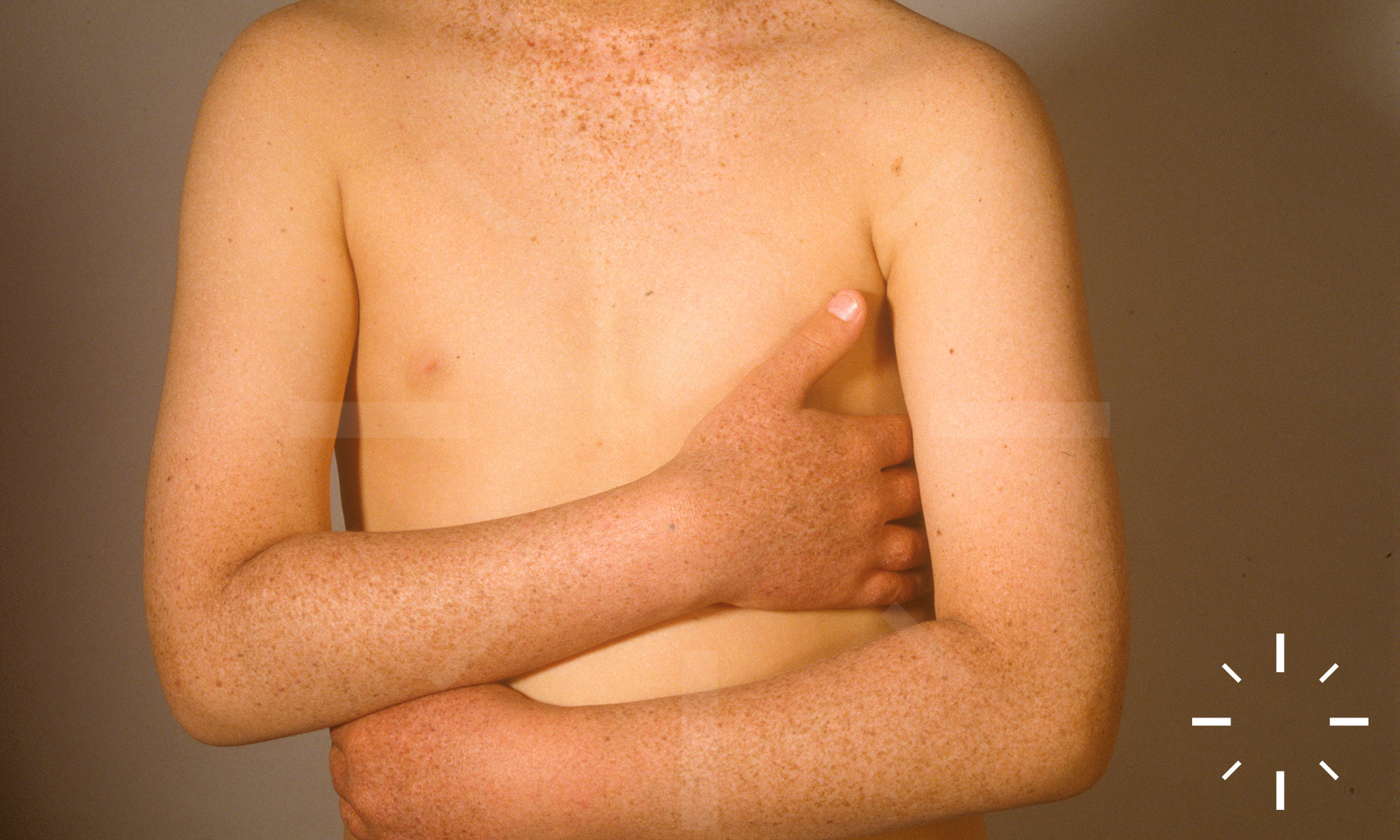
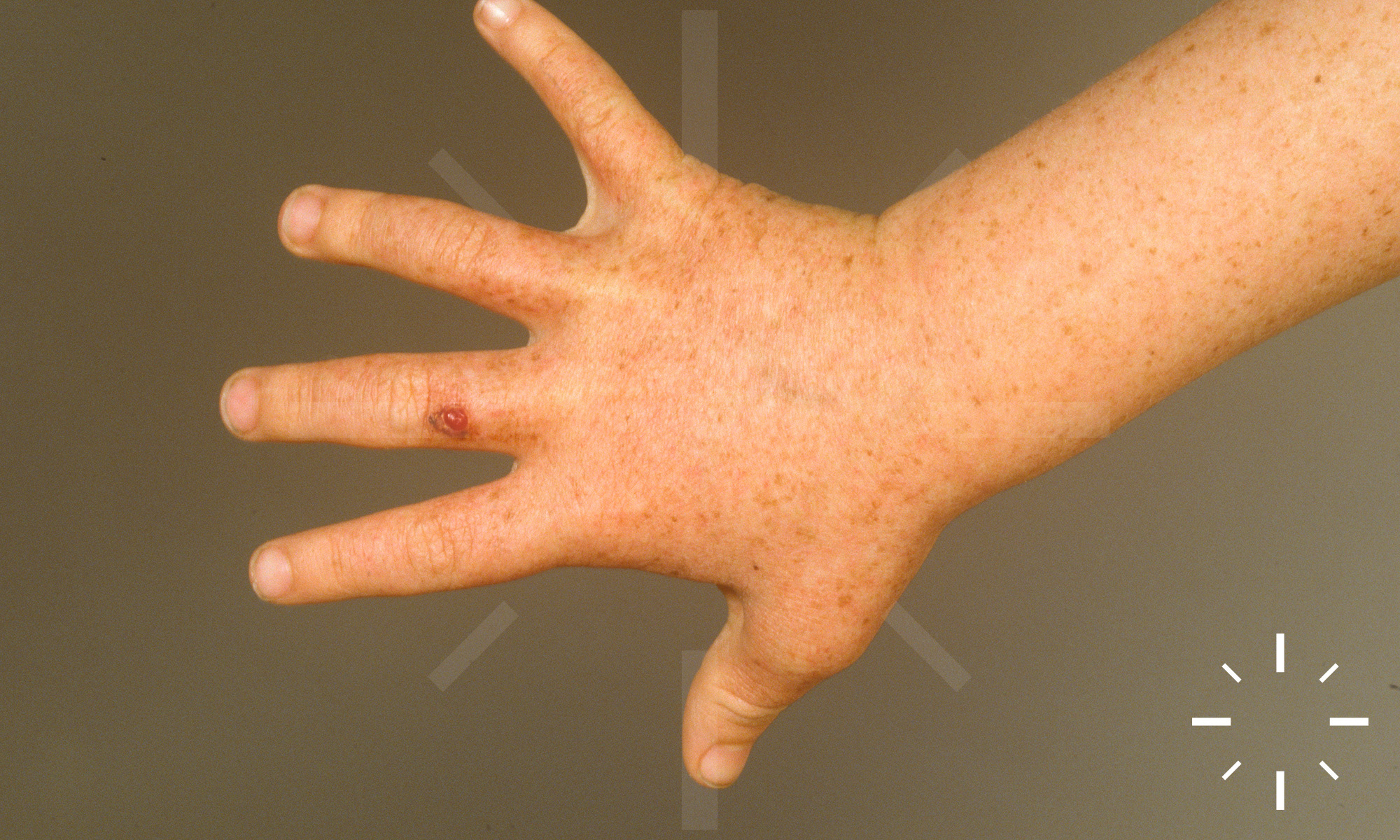
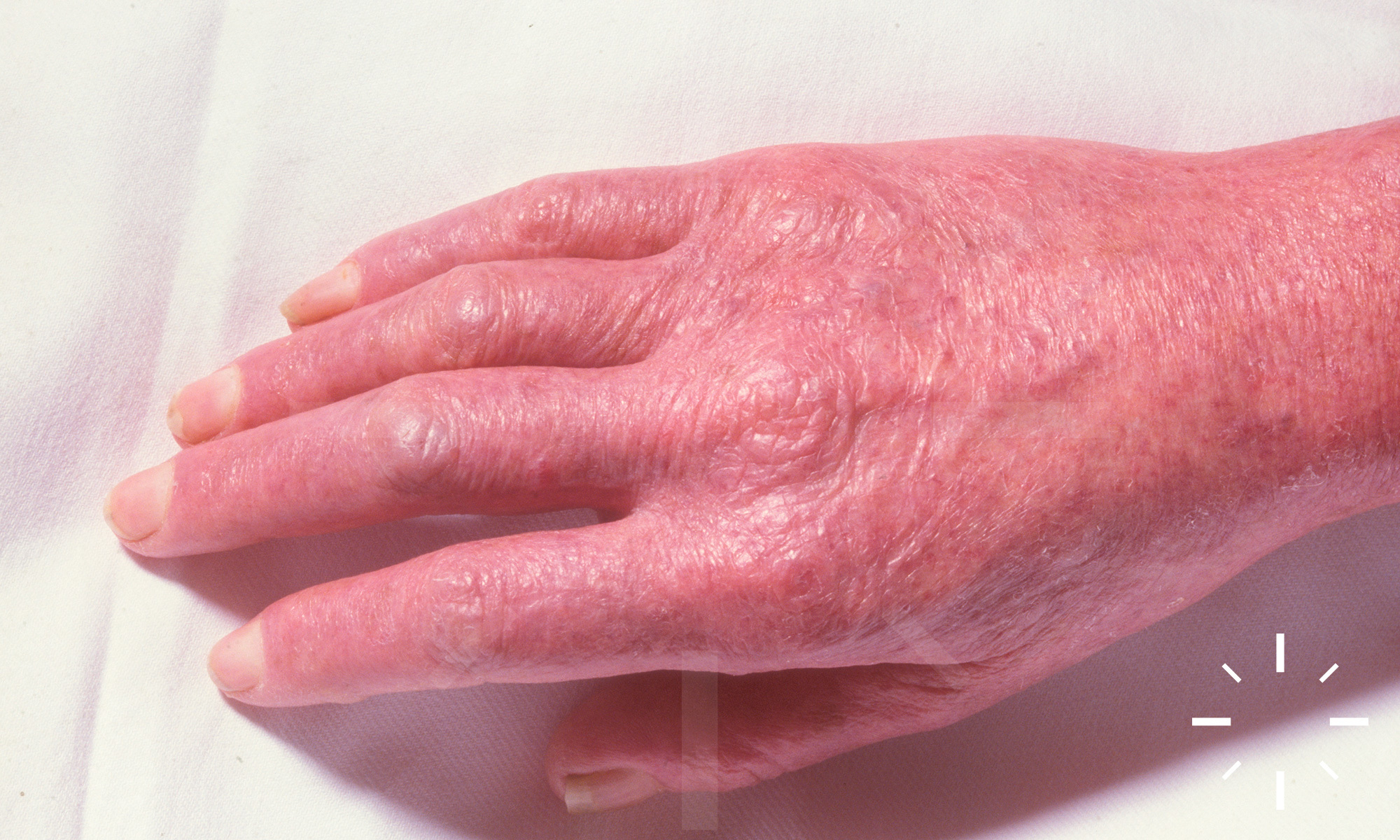
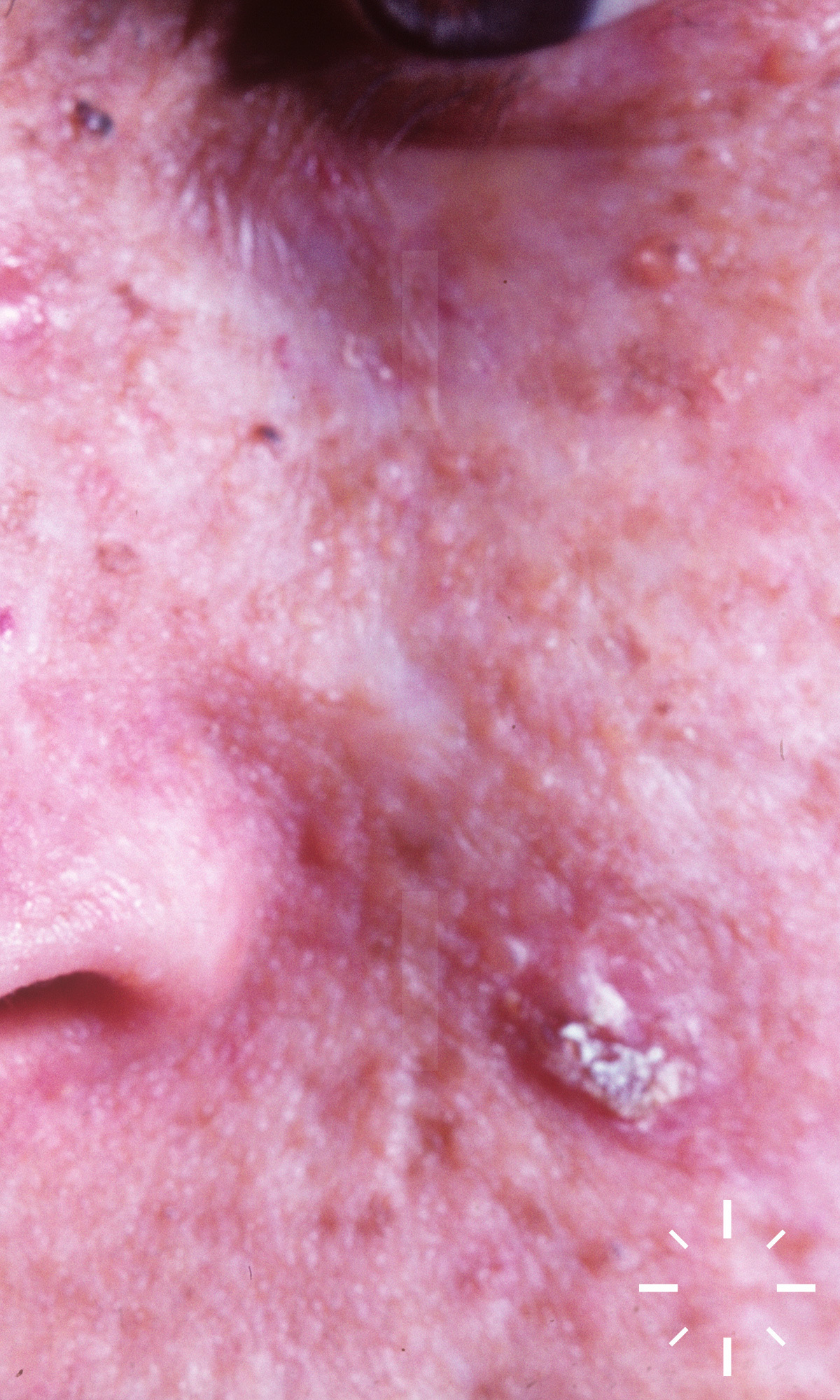
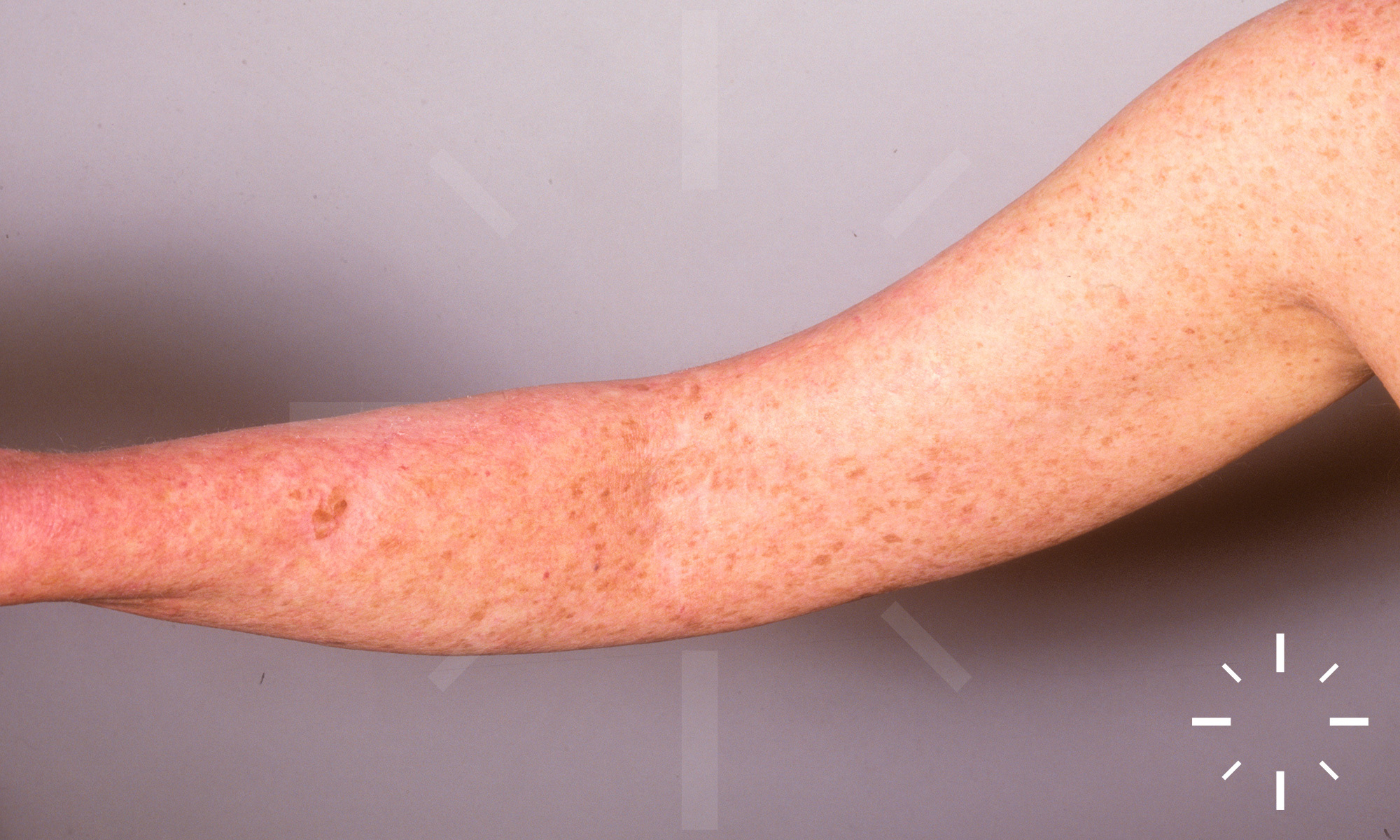
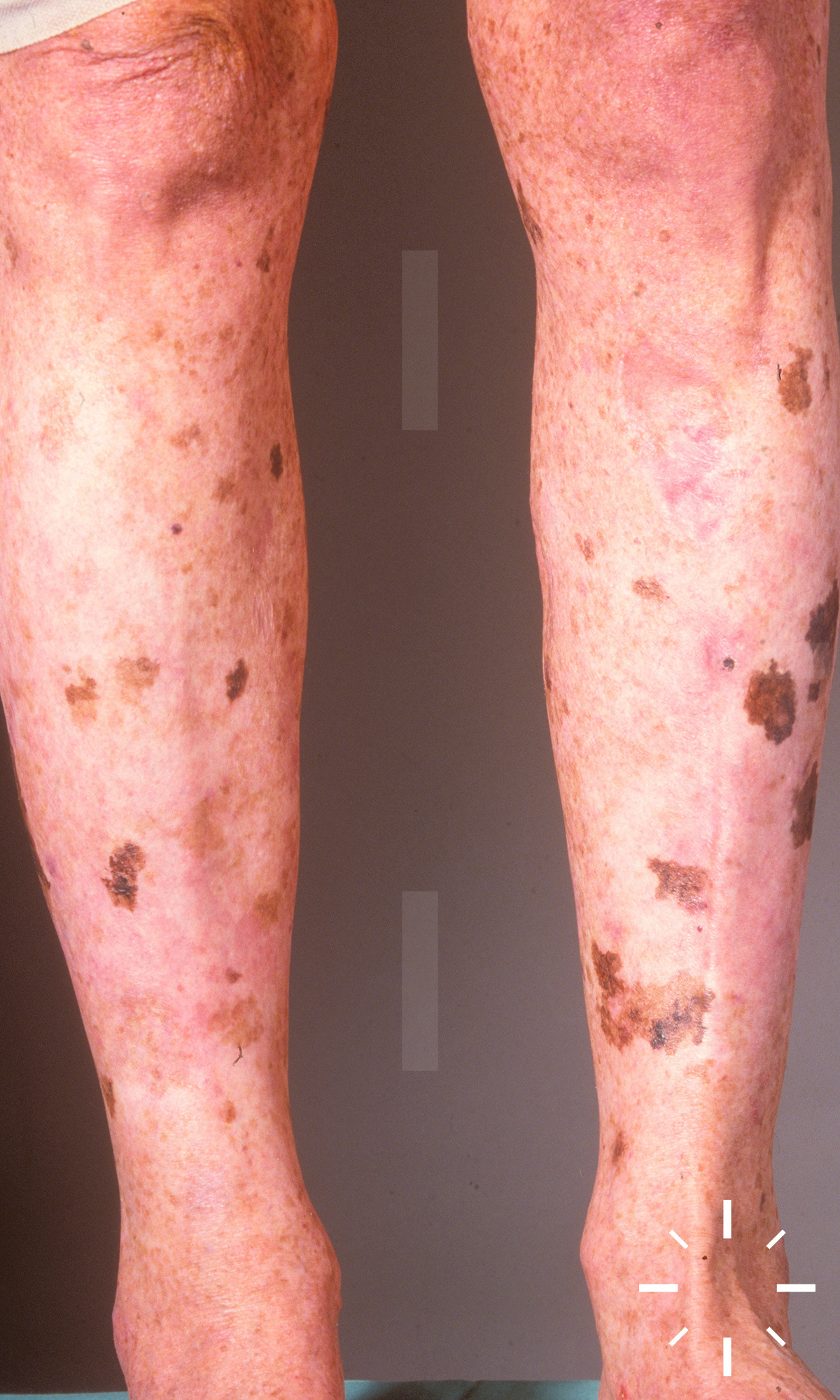
Xeroderma pigmentosum
Last Updated: 2023-07-07
Author(s): Anzengruber F., Navarini A.
ICD11: LD27.1

1/11